
If you know us, then you know that clean air and water is our passion in life. But what happens when you cannot prove to the regulatory authorities that your air and water is clean?
Cleanrooms and monitoring systems generate a lot of data. In an 8 hour shift, as many as 5,000 individual count samples can be generated. That is a lot of data to collate and track. But without proper measures, data can become corrupted and the cleanroom could be considered compromised.
That is why we need to take steps to certify data integrity.
The US Food And Drug Administration (FDA) defines data integrity as the “completeness, consistency, and accuracy of data. Complete, consistent, and accurate data should be attributable, legible, contemporaneously recorded, original or a true copy, and accurate (ALCOA).” In a cleanroom, data integrity certifies to the regulatory authorities that the cleanroom is following best practices and producing safe products.
While maintaining the highest level of data integrity might seem daunting in a cleanroom that generates thousands of pieces of data every hour, it is possible. First, you must understand a few of the guidelines and regulations surrounding cleanroom data integrity, including 21CFR11 and GAMP. Next, you should implement a paperless system that does the hard work for you.
But first, let’s start with the basics.
What Is 21CFR11?
21CFR11 refers to the 11th part of the FDA’s Title 21 Code of Federal Regulations. This is the portion of the regulations that give guidance on how to handle data that has to be submitted to the FDA. This measure allows for the use of digital technology in the handling of data, as it outlines how to ensure the utmost confidence in your data integrity.
The FDA introduced 21CFR11 in 1996. Since then, there have been numerous changes to Part 11 to keep pace with technological changes. This is not a stagnant document, but a statute that evolves with the industry needs since its creation. The original purpose of Part 11 was to allow pharmaceutical companies more agility in the production of highly regulated products. Instead of chasing physical signatures and collating paper documents for FDA audits, organizations have been able to digitize much of the process.
Part 11 balances the industry’s need to quickly bring products to the market with the requirement of the highest level of authentication and control.
To help organizations comply with 21CFR11 and maintain their data integrity, the FDA introduced the concept of ALCOA in the 1990s. They would later go on to release ALCOA+, which is now considered standard practice to maintain data integrity for numerous organizations, including the FDA and GAMP.
What Is ALCOA+?
ALCOA stands for Attributable, Legible, Contemporaneous, Original, and Accurate. ALCOA+ goes on to include that data must also be Complete, Consistent, Enduring, and Available. These are the basics of maintaining a high level of data integrity compatible with the high standards of the regulatory authorities all over the world.
Attributable: It must be clear who recorded the date, with a signature and date.
Legible: The data must be clearly able to be understood without unexplained symbols.
Contemporaneous: Data should be recorded immediately upon its generation.
Original: The data should be the original or a certified copy.
Accurate: Data should accurately reflect the situation or observation.
Complete: Records should leave nothing out – include testing and re-testing.
Consistent: Time stamps should come in the expected sequence. Across the board, data generation should be the same.
Enduring: Data should only be recorded using invalidated electronic systems (or, if unavailable, controlled worksheet in laboratory notebooks).
Available: For the lifetime of the record, it needs to be readily available for review and audits.
Following these guidelines, you should be able to maintain the highest level of data integrity possible and comply with standards set forth by regulatory agencies, such as the FDA and GAMP.

What Is GAMP5?
GAMP stands for Good Automated Manufacturing Practices. It is both a subcommittee of the International Society for Pharmaceutical Engineering (ISPE) and a set of guidelines for pharmaceutical cleanrooms. GAMP5 specifically refers to the section of the guidelines that cover how to design a cleanroom Environmental Monitoring System (EMS) to maintain data integrity and produce a safe product.
While GAMP5 is technically guidelines for pharmaceutical organizations, the information put forth is valuable for anyone using a cleanroom.
GAMP5 uses a V model to demonstrate the steps you should take to implement your EMS. This is an exhaustive process to help you address all issues at the design level, instead of later on during the application phase. While it might feel overly thorough while going through it, you’ll be thankful for the process when the implementation is much more seamless and fewer problems arise.
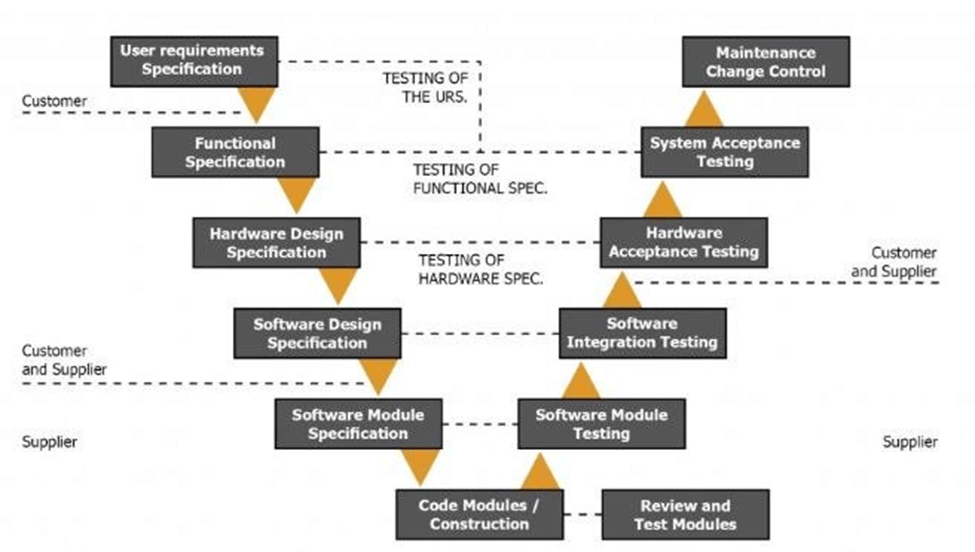
While 21CFR11 regulates data integrity, GAMP5 shows you how to design for data integrity.
Both are important statutes for you to familiarize yourself with and are considered around the world.
What Does An EMS Have To Do With 21CFR11 and GAMP5?
Both 21CFR11 and GAMP5 iterate the importance of data integrity. Without that, your cleanroom won’t be able to pass audits or receive a certification. The key to your data integrity is using an EMS that has been designed around these guidelines and regulations with data integrity and quality at the forefront of the innovation.
That is what we do here at Lighthouse.
Our Environmental Monitoring Systems are designed to follow best practices set forth by GAMP5 and 21CFR11 to maintain data integrity. We build these guidelines into the EMS and allow for further customization on your part. With our powerful software integrations, you can pull reports easily and prove data integrity.
Moreover, we want to make sure your system is validated from the start, which is why we offer a unique cleanroom monitoring system validation. Want to know more? Contact us today.